

Agosto/2014
Carlos Fioravanti | Revista Pesquisa Fapesp
De Monte Santo, BA

A via-sacra de Monte Santo: local de peregrinação que recebe milhares de pessoas todo ano. © EDUARDO CESAR
José de Andrade Pereira é um homem de fibra. Em 2004, ele levou o filho mais velho, que aos três anos era muito baixo, tinha dedos curtos, cabeça grande e dificuldade de fala – e mais uma vez estava com forte dor de ouvido –, a um posto de saúde de Monte Santo, interior da Bahia. O médico lhe disse que, além de cuidar da dor de ouvido, não poderia fazer mais nada diante de uma doença que não conhecia e que ele deveria apenas esperar o menino morrer. Pereira reagiu: “Esperar é o que não vou fazer, nunca!”. Ele fez a viagem de seis horas até Salvador e perguntou a um porteiro do Hospital Universitário Professor Edgard Santos quem ele deveria procurar para tratar de um menino como aquele. Os médicos examinaram o menino e depois o irmão de 11 meses, na viagem seguinte, e concluíram que os dois tinham mucopolissacaridose tipo 6, uma doença rara de origem genética então sem tratamento. Pereira alertou: “Tem outras crianças assim por lá”. Sua visão de mundo mudou a história desta cidade do sertão baiano.
Monte Santo foi um acampamento para as tropas do governo que lutaram na guerra de Canudos. A praça principal exibe uma escultura em madeira de Antonio Conselheiro, o beato que liderou os sertanejos vistos como opositores da república nascente. Apontada para a escultura há uma matadeira, canhão usado nas batalhas em que morreram 25 mil revoltosos e 5 mil soldados. Nos últimos anos Monte Santo tem sido o palco de outras batalhas: a identificação, o tratamento e a prevenção de doenças genéticas raras, que começaram a ser reconhecidas a partir da indicação de Pereira. Antes as crianças com doenças como a mucopolissacaridose permaneciam em suas casas. Seus pais achavam que nada mais poderia ser feito.

Os ex-votos da capela no final da trilha de pedra. © EDUARDO CESAR
Médicos e pesquisadores de Salvador, Rio de Janeiro e Porto Alegre foram a Monte Santo pela primeira vez em 2006 e se espantaram com a diversidade de doenças raras que viam em um só lugar. Já diagnosticaram 13 pessoas com mucopolissacaridose tipo 6, uma proporção 240 vezes maior do que a média nacional, 84 com deficiência auditiva de possível origem genética, 12 com hipotireoidismo congênito, nove com fenilcetonúria, que pode causar deficiência intelectual se não tratada, quatro com osteogênese imperfeita, marcada pela extrema fragilidade dos ossos, e quatro com síndrome de Treacher Collins, que prejudica a formação dos ossos do crânio.
Acredita-se que os casamentos entre parentes, antes muito frequentes, possam ter favorecido o surgimento de doenças físicas e mentais de origem genética. Muitas pessoas se casavam sem saber que tinham ascendentes próximos em comum. José Pereira e sua esposa, Júlia Isaura dos Santos Pereira, souberam que eram parentes só há poucos anos, ao reconstituírem a genealogia da família com os pesquisadores de Salvador, e enfim entenderam por que tinham ouvido falar de tios com a mesma doença dos dois filhos mais velhos. Talvez as raízes mais profundas desses problemas estejam na própria história do lugar. Vários relatos do historiador baiano José Calasans indicam que o município hoje com 52 mil moradores – espalhados em 47 povoados ao redor do núcleo urbano – foi um centro de convergência de pessoas doentes em busca dos milagres do Conselheiro, que reforçou a fama religiosa do lugar. Foi ele quem reformou as capelas ao longo da via-sacra, um caminho íngreme e sinuoso de pedras, com 2,8 quilômetros (km) de extensão, que termina em uma capela erguida no alto do morro em 1786 por um padre italiano. Todo ano, milhares de romeiros sobem o caminho de pedras, às vezes de joelhos ou com uma pedra na cabeça, para pagar promessas. Por serem geralmente pobres, os doentes, curados ou não, seus familiares e romeiros podem ter tido dificuldade para voltar para suas terras de origem ou preferido ficar na região.

Monte Santo ao anoitecer: palco de batalhas históricas. © EDUARDO CESAR
Como deve haver mais pessoas ainda não diagnosticadas, a médica geneticista Angelina Acosta, professora da Universidade Federal da Bahia (UFBA), subiu ao auditório da Câmara de Vereadores no início da tarde do dia 10 de julho e expôs a médicos e políticos seu plano de fazer um censo da saúde de toda a população. “Somos de universidades, mas trabalhamos com vocês todos, para que nosso trabalho tenha uma aplicação prática”, ela afirmou. A secretária municipal da Saúde, Itácia Macedo de Andrade Silva, acompanhava tudo com atenção. “Quero fazer algo pela minha terra”, disse ela, explicando por que voltou à cidade depois de estudar enfermagem em Salvador. Na manhã seguinte, a equipe coordenada por Angelina e por Kiyoko Sandes conversou com 80 agentes comunitários da saúde que vão visitar os povoados em busca de outros casos, a partir deste mês. O diálogo resultou em indicações de pessoas que ainda não haviam sido examinadas e na formação de um comitê para acompanhar o censo e o tratamento. José Pereira era um dos integrantes.
As doenças raras formam um mundo de sofrimento, solidão, fantasias e culpa, que começa a ser examinado publicamente. “O Sistema Único de Saúde [SUS] reconheceu que as doenças raras devem ser tratadas”, comenta Clarice Alegre Petramale, diretora do departamento de gestão e incorporação de tecnologias do Ministério da Saúde. A política nacional de atendimento médico às pessoas com doenças raras – definidas como qualquer enfermidade apresentada por até 13 pessoas em cada grupo de 20 mil indivíduos – está em vigor desde maio deste ano. Os grupos de doenças prioritárias para atendimento devem ser anunciados até o fim deste ano.
 Para a maioria das doenças raras não há medicamentos específicos, apenas tratamento de apoio, como fisioterapia e fonoaudiologia. Quando existe medicação, é geralmente importada e obtida por meio de decisões judiciais. “Os remédios são caros e muitas vezes de eficácia incerta, porque não passaram por todos os testes rigorosos de avaliação, já que foram testados em um número muito pequeno de pacientes”, diz Magda Carneiro-Sampaio, diretora do Instituto da Criança da Universidade de São Paulo (USP). “E os medicamentos podem ser indicados já em estágios avançados das doenças, quando não funcionam tão bem. É uma situação mal equacionada, mas não é só no Brasil.” Maíra Catharina Ramos, da Universidade de Brasília, calculou que a compra voluntária pelo governo de apenas um medicamento usado para tratar mucopolissacaridose implicaria uma economia de R$ 50 milhões, em relação ao que é gasto para atender as decisões judiciais, que obrigam o governo a comprar os remédios. Estima-se que 13 milhões de pessoas no país tenham alguma doença rara, das quais, em todo o mundo, já foram descritos 6 mil tipos, a maioria de origem genética.
Para a maioria das doenças raras não há medicamentos específicos, apenas tratamento de apoio, como fisioterapia e fonoaudiologia. Quando existe medicação, é geralmente importada e obtida por meio de decisões judiciais. “Os remédios são caros e muitas vezes de eficácia incerta, porque não passaram por todos os testes rigorosos de avaliação, já que foram testados em um número muito pequeno de pacientes”, diz Magda Carneiro-Sampaio, diretora do Instituto da Criança da Universidade de São Paulo (USP). “E os medicamentos podem ser indicados já em estágios avançados das doenças, quando não funcionam tão bem. É uma situação mal equacionada, mas não é só no Brasil.” Maíra Catharina Ramos, da Universidade de Brasília, calculou que a compra voluntária pelo governo de apenas um medicamento usado para tratar mucopolissacaridose implicaria uma economia de R$ 50 milhões, em relação ao que é gasto para atender as decisões judiciais, que obrigam o governo a comprar os remédios. Estima-se que 13 milhões de pessoas no país tenham alguma doença rara, das quais, em todo o mundo, já foram descritos 6 mil tipos, a maioria de origem genética.
De norte a sul
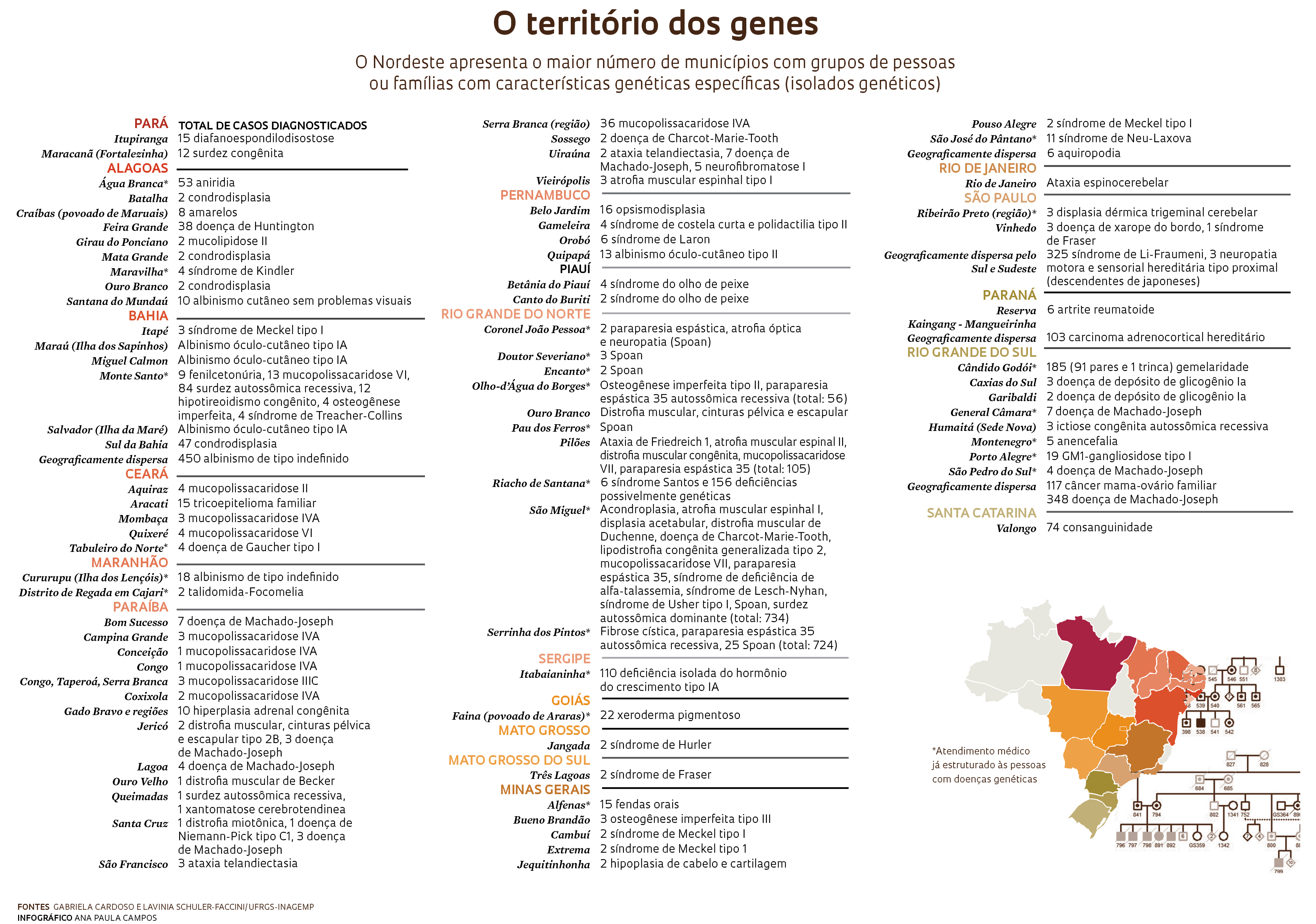
O trabalho dos pesquisadores acadêmicos com os profissionais da saúde locais em busca de outros moradores com doenças incomuns coloca Monte Santo na linha de frente do Censo Nacional de Isolados (Ceniso), organizado pelo Instituto Nacional de Genética Médica e Populacional (Inagemp). Em abril de 2014, um artigo publicado na revista Genetics and Molecular Biology apresentou um dos resultados do Ceniso: um levantamento nacional dos municípios com grupos de pessoas ou famílias com doenças genéticas. Uma versão atualizada desse mapeamento, apresentada na página anterior, reúne 81 municípios, onde vivem 4.136 pessoas com características genéticas específicas, os chamados isolados genéticos. Nem sempre são doenças. O município gaúcho de Cândido Godói, por exemplo, apresenta um número extraordinário de gêmeos. A equipe da médica geneticista Lavínia Schuler Faccini, professora da Universidade Federal do Rio Grande do Sul (UFRGS) e membro do comitê coordenador do Inagemp, identificou 91 gêmeos duplos e um triplo nascidos entre 1959 e 2000 no município, hoje com 6 mil moradores. Entre 1994 e 2006, 2% das crianças nascidas em Cândido Godói eram gêmeas, o dobro da média nacional. Em um dos distritos a taxa de gêmeos chegou a 10%.
Algumas doenças genéticas se manifestam na idade adulta, mesmo depois dos 40 anos de idade, como a ataxia de Machado-Joseph, que causa a perda progressiva do equilíbrio, dos movimentos e da fala. Lavínia, com sua equipe, que faz o aconselhamento genético dos familiares de quase 400 pessoas com ataxia em Porto Alegre, tem observado que o diagnóstico em geral tardio costuma gerar angústia e culpa, porque as pessoas já podem ter tido filhos ou netos com as mutações causadoras da doença.
Há doenças de alcance regional, como a síndrome de Li-Fraumeni, uma forma genética hereditária de predisposição ao câncer, já identificada em 325 pessoas dos estados do Rio de Janeiro, São Paulo, Paraná, Santa Catarina e Rio Grande do Sul. Um estudo recente registrou uma prevalência alta, de 0,27%, da mutação causadora da doença em 171 mil bebês de Curitiba examinados, indicando que essa doença, em alguns lugares, não é rara e necessitaria de um acompanhamento intensivo, principalmente com as pessoas de risco mais alto.

Primas com fenilcetonúria controlada: Raíra Anielli Carvalho Silva, entre a mãe, Eliana Batista Carvalho, e o irmão, Ranieri Carvalho Silva (esquerda), e o avô de 92 anos, José Lopes de Carvalho, e o pai, José Nildo Andrade Silva. © EDUARDO CESAR
O médico Eduardo Enrique Castilla, pesquisador da Fiocruz do Rio de Janeiro e um dos idealizadores do censo, acredita que o total de municípios já identificados com doenças genéticas raras no Brasil deve representar apenas 20% do previsto. A lista não para de crescer porque os pesquisadores continuam encontrando indicações de outros lugares ainda não mapeados. Em junho deste ano, o geneticista Carlos Menck, da USP, foi a Miracatu, cidade de 30 mil habitantes a 130 km de São Paulo, e identificou quatro pessoas de uma mesma família com xeroderma pigmentoso, já diagnosticada em 22 dos moradores de um povoado no interior de Goiás. Embora a doença seja a mesma, as mutações que a causam, os genes e os cromossomos atingidos são diferentes nas pessoas dos dois estados.
Desde 2013, a equipe da médica geneticista Denise Cavalcanti, da Universidade Estadual de Campinas (Unicamp), identificou novos clusters, outro nome para os isolados genéticos, de diferentes displasias esqueléticas, doenças que prejudicam o crescimento dos ossos. Os clusters estão em cinco municípios dos estados do Ceará, Alagoas, Pernambuco e São Paulo. Um deles, identificado em colaboração com uma médica geneticista de Fortaleza, impressiona pelo número de pessoas diagnosticadas até agora – 27, de 22 famílias. Dispersas em pelo menos 10 cidades pequenas do interior do Ceará, elas têm uma doença bastante rara chamada picnodisostose, a mesma que determinou a baixa estatura do artista francês Henri de Toulouse-Lautrec. Denise, com seu grupo, trabalha agora para identificar o possível local de origem da mutação que causa a doença, o chamado efeito fundador, no Ceará.

Camilly Vitória de Souza Andrade, entre o pai, José Armando Moraes de Andrade, e a mãe, Cremilda Maria de Souza Andrade. © EDUARDO CESAR
“Você não imagina como é importante para as mães saber o nome da doença dos filhos, mesmo que não exista tratamento, porque aí param de andar de um médico para outro”, diz Denise. Um dia ela recebeu uma carta de uma mulher de Belém que agradeceu pelo diagnóstico de um filho e comentou: “Era angustiante não termos um diagnóstico, era como andar no escuro ou sem rumo”. Quando conhecem o nome das doenças, as mães “voltam a apostar nos filhos”, observa Isabella Queiroz, professora da Escola Bahiana de Medicina e psicóloga da Apae de Salvador que atende as famílias com doenças genéticas de Monte Santo. “Já fizemos mais de 200 sessões de aconselhamento genético.” Nesses encontros, a equipe médica explica que as doenças genéticas hereditárias resultam da transmissão de genes com alterações (ou mutações), porque apenas o casamento entre parentes não é a única explicação. Ao ver um mapa genético da família relacionando as pessoas saudáveis e doentes, uma mulher entendeu a seu modo o que acontecia: “Juntou uma manchinha minha com outra de meu marido e nasceu prejudicado, é isso?”. É também quando transbordam a culpa por ter tido filhos doentes, o desamparo e a raiva. Um dos homens questionou quem o atendia: “Não posso mais ter filhos? Então estou condenado?”. A equipe de aconselhamento genético sabe que deve expor os riscos de doenças hereditárias sem intervir sobre a escolha dos casais de terem filhos.
Às vezes, casais mais jovens, antes de ter filhos, procuram voluntariamente o serviço médico para detectar eventuais mutações prejudiciais, indicando que o número de casos de algumas doenças genéticas pode cair nos próximos anos. O hábito de casar com primos – muito mais frequente nos países muçulmanos do que no Brasil – é que talvez seja mais difícil de mudar, porque tem sido adotado há muito tempo, como forma de manter as terras em família ou por preferências pessoais. Quando visitaram Tabuleiro do Norte, município a 200 km de Fortaleza com alta prevalência de uma doença metabólica conhecida como síndrome de Gaucher, os pesquisadores ouviram os homens dizerem que as mulheres da capital eram boas para namorar e as de Tabuleiro, boas para casar. Permanecer atrelado ao próprio lugar é que poderia, portanto, causar o índice elevado de doenças genéticas. Um padre de Monte Santo, conta-se, ofereceu bicicletas para os rapazes buscarem noivas em outros lugares, mas ninguém aceitou.

Livro de casamentos da paróquia de Monte Santo em 1938: no caso de primos, o vigário tinha de dar “dispensa de impedimento”. © EDUARDO CESAR
“Muita coisa mudou”, observa Maria Olívia Sousa Costa, secretária da Saúde de Monte Santo de 2008 a 2011. “Os médicos se tornaram responsáveis pela identificação de doenças raras, e as mães ganharam consciência de seus direitos à saúde e reclamavam quando faltava infusão.” Infusão é um preparado com a enzima arilsulfatase B, que as crianças com mucopolissacaridose tipo 6 não produzem e é aplicada em sessões de quatro horas, uma vez por semana. Durante anos os pais levavam as crianças a Salvador para receberem a medicação, desde 2011 aplicada em um anexo do hospital da cidade. Nem tudo mudou, porém, porque nem sempre há medicação. “Tem de cobrar sempre, mas não me acanho, não”, diz Pereira. Seus dois filhos começaram a ser tratados em 2008 em Salvador, quatro anos depois do diagnóstico, já que a enzima não tinha sido ainda liberada para uso no Brasil. A doença não pôde ser revertida. Hoje Jorge tem 17 anos e seu irmão Sidney, 14, ambos com menos de um metro de altura e com dificultades de movimento. Vitor Gonzaga Andrade, filho de Alaíde Gonzaga Andrade, prima de Pereira, começou a receber a medicação aos dois anos e hoje, aos seis, corre como um menino saudável.
Cremilda Maria de Souza Andrade se sentiu desarvorada ao saber que a filha Camilly tinha fenilcetonúria, diagnosticada um mês depois de nascer. Foi para Salvador e começou o tratamento, que consiste em uma dieta rigorosa, sem nenhuma proteína que contenha fenilalanina, que o organismo não consegue processar. “Hoje estou feliz”, diz Cremilda, mostrando com orgulho os cadernos da filha, agora com 11 anos e sempre atenta à dieta. Camilly é prima de Raíra Anielli Carvalho Silva, de 12 anos, que também tem fenilcetonúria, detectada logo após nascer e controlada por meio da alimentação. No mesmo povoado de Raíra, porém, moram dois irmãos, hoje com 40 e 27 anos e a mesma mutação das meninas. Como o teste do pezinho ainda não era comum na região quando eles nasceram, a doença deles foi detectada muito depois, não foi controlada e causou os distúrbios mentais que os impedem de sair de casa.
Pé na estrada
Para fazer um trabalho abrangente, os pesquisadores têm de sair do laboratório, colocar uma roupa de estrada, viajar para lugares inimagináveis, conhecer os hábitos e os silêncios dos moradores do interior do país e buscar informações em outros espaços. Para localizar pessoas com maior risco de doenças genéticas, Angelina Acosta e sua equipe consultaram os livros de casamentos, batizados e mortes na paróquia de Monte Santo desde 1831, e refizeram a história de 1.419 famílias. Depois de se perderem, encontraram o caminho das pedras, valioso até mesmo para os historiadores, ao verem que as mulheres ganhavam sobrenome apenas depois de casadas, os casais registravam as filhas com o sobrenome da mãe e o filho com o sobrenome do pai, além de apelidos usados como nomes – um homem conhecido como Santana na verdade chamava José da Silva. Em Monte Santo, nomes e sobrenomes se repetiam em um labirinto como o da família Buendía em Cem anos de solidão, cujos integrantes viviam casando entre si e depois temiam ter filhos com rabo de porco.
A publicação de artigos científicos ainda é importante, mas não é a prioridade, porque há “uma obrigação moral” de relatar as descobertas primeiramente às comunidades estudadas, enfatiza Castilla. Em uma manhã de sábado, Lavínia e sua equipe se puseram à frente de 200 pessoas no salão de festas da igreja de Cândido Godói para explicar o motivo do número elevado de gêmeos, muitos deles na plateia: era uma provável consequência de uma variação do gene da proteína p53, que poderia favorecer o desenvolvimento de dois embriões por gestação. Durante décadas se acreditou que os gêmeos eram um efeito da água supostamente especial do município.
Artigos científicos
CASTILLA, E. E. & SCHULER-FACCINI, L. From rumors to genetic isolates.Genetics and Molecular Biology. v. 1, n. 37, p. 186-93. 2014.
TAGLIANI-RIBEIRO A. et al. High twinning rate in Cândido Godói: a new role for p53 in human fertility. Human Reproduction. v. 27, n. 9, p. 2866-71. 2012.
MACHADO, T. M. B. et al. Types of marriages, population structure and genetic disease. Journal of Biosocial Science. v. 45, n. 4, p. 461-70. 2013.
Fonte: Revista Pesquisa Fapesp
Leia em HCSM:




